 hotline服务热线:010-61006450
hotline服务热线:010-61006450
案例剖析 | pbpk模型在不同晶型原料药制剂研发中的可行性-凯发k8旗舰厅
材料
☆ 参比制剂:速释片剂20mg,已上市
☆ 原料药:结晶形式
☆ 仿制药:sandoz公司开发,原料药晶型为与二氧化硅形成的无定型态,规格20mg,速释片剂
体外溶出和理化性质
☆ 溶出方法:ph6.5 空腹状态下的人工胃液(fassif)介质、桨法、1000ml、75rpm。 晶型(参比制剂)溶解度为0.07mg/ml;两种原料药在fessif介质中的溶解度分别为0.019mg/ml和0.011mg/ml
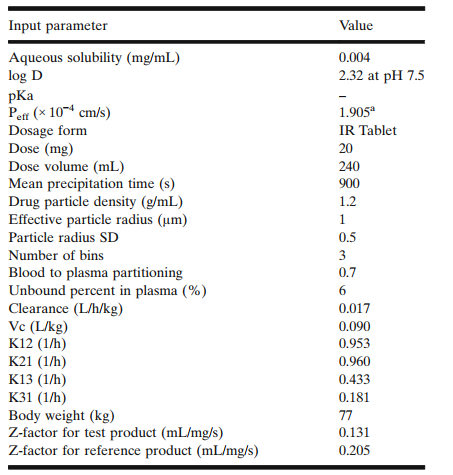
图1 不同晶型原料药理化性质和药动学参数
模型方法
将原料药内部、文献数据、处方性质(图1),以及体内药动学参数(参考参比制剂空腹时的药动学参数)等数据输入至gastroplustm软件中,并调整小肠和结肠的流体体积,优化模型。建立模型也经过了内部验证(参比制剂结果)和外部验证(仿制药结果)。
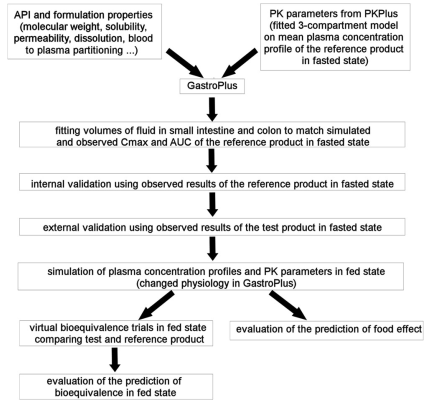
图2 模型建立的流程
该模型建立流程为:
-
将原料药性质如分子量、溶解性、渗透性、溶出和全血血浆及pk参数(参考参比制剂空腹平均血药浓度,3房室模型)输入至gastroplus软件中。
-
优化小肠和空肠流体体积,使模型中cmax和auc值拟合
-
采用空腹条件下参比制剂的结果进行内部验证
-
采用空腹条件下仿制药的结果进行外部验证
-
模拟餐后条件下血浆浓度(在软件中改变生理条件)
-
餐后条件下进行虚拟be,预测食物影响
-
评估餐后生物等效性预测
① z-factor模型
软件中体外溶出采用z-factor模型;pk模块的参数设定,参考空腹参比制剂的pk模型,模拟结果为三房室模型拟合结果较好,生物利用度40%;采用已确定的肝脏清除率、全血血浆比例、肝脏血流速度,计算首过效应。空腹状态下肝脏首过效应为2.0%,餐后状态下肝脏首过效应为1.5%。
② 人体生理模型
对于软件中构建的人体生理模型,小肠和空肠体液体积,将小肠中的默认值由40%优化为10%,将空肠的体液体积由23%优化为0.5%。当模拟餐后状态,采用软件中默认的餐后模型(human-physiology-fed, opt logd model sa/v 6.1 asf model)。
☆ 单剂量空腹模拟结果:
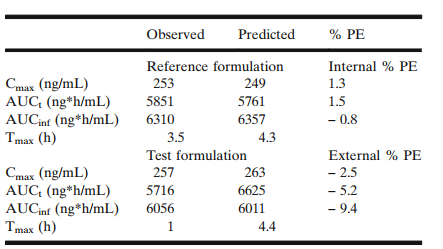
图3 空腹状态下单剂量观测值和预测值对比
采用参比制剂的pk和体外数据,建立模型,并采用该模型,进行内部验证,pe小于10%,符合fda的指导原则。根据该原则,可以建立ivivc。采用ivivc模型,模拟每个处方,要求模拟的结果和实测值的pe不超过15%,该模型可以用于模拟仿制药的血药浓度。
仿制药和参比制剂,因z-factor值不同,模拟出在体内的溶出速率也不同。采用该模型进行虚拟be,结果表明仿制药和参比制剂在空腹状态下生物等效,cmax的90%置信区间为91.262%-122.35%,auc的90%置信区间为91.086%-119.68%。
☆ 餐后模拟结果:
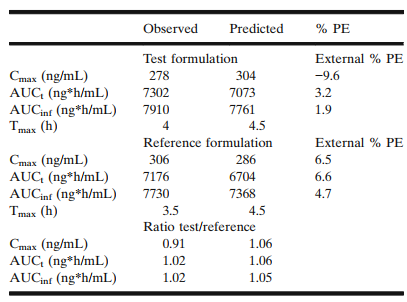
图4 餐后状态下单剂量观测值和预测值对比
上述开发的模型用于预测仿制药和参比制剂在餐后状态下的平均血药浓度,预测的cmax和auc均比空腹条件下偏高,原因为在体内溶出速率提高,由于胃肠道转运时间提高及胃的流体体积增加。因药物的溶解性不存在ph依赖性,不同的ph不影响溶出速率。
采用上述模型,32例受试者进行虚拟be试验,仿制药和参比制剂的cmax和auc均为80.00%-125.00%范围内,两者具有生物等效性。外部验证的模型pe值小于10%。
☆ 餐后模拟结果:
gastroplus软件采用pbpk模型预测药物在体内的adme(吸收、分布、代谢、消除)过程。该模型根据体外的数据,用于处方早期开发,基于不同处方的体外的性质,预测药物在体内的血药浓度,为优化处方做决策;比较仿制药和参比制剂的差异,通过参数敏感性分析和确定关键参数,建立ivivc(体内外相关性),判断溶出标准,申请豁免,进行虚拟be试验等。
模型假设
建立模型过程中,相应进行了假设,如假设参比制剂原料药为微粉化,仿制药的无定型原料药在储存过程中无变化,两个原料药的水溶解性一致。在模型中加入原料药和制剂在生物相关性介质中的粒度和溶解度差异,将生物相关性介质fassif输入模型中,仿制药因为溶出较高,具有较高的z-factor值。
该模型不能预测仿制药在空腹状态下具有较短的tmax(1h),可能由于一些个吸收较快导致。总体来说,观测值tmax为范围,仿制药和参比制剂的tmax观测值分别为0.5-6.0h和0.75-5.0h,预测值分别为4.4h和4.3h。仿制药和参比制剂的观测值tmax的平均值分别为2.27h和2.41h。表明仿制药和参比制剂观测值和预测值,两个制剂趋势一致。
该模型也假设胃中无吸收,对于大多数的化合物,胃中的吸收可以忽略,但并不包含在gastroplustm默认模型中。模型开发过程中,增加了胃吸收,但tmax仍没有提高。说明药物进入十二指肠后,依然存在药物的溶解过程,因为溶出是缓慢并且是不完全的,限制了药物的吸收,当药物最大程度上溶解,会得到最高的血药浓度。该过程发生在4h,而体内,在一些个体中,溶解性会更快,tmax较短。
药动学参数采用空腹的be数据,pk-plustm模块,拟合3房室模型,选择3房室模型为pk药动学参数。因bcsii类药物,无iv(静脉)数据,并且无溶液在体内的吸收数据,也没有绝对生物利用度。因无未溶解药物的总量、无首过效应数据和药物肝肠循环数据。基于质量平衡研究,药物服用后,36%剂量在尿液中检测到,61%剂量在粪便中检测到。基于该数据,假设生物利用度至少为40%。考虑到较低的生物利用度,模型对于溶出更为敏感。
优化模型后,小肠和空肠的体液体积从40%和10%,优化为23%和0.5%,使观测值和预测值相拟合。上述参数也用在餐后状态下。
对于不同晶型食物的影响
对于预测不同晶型的药动,需要将不同晶型的数据纳入至模型中去,如水溶性、fassif和fessif的溶解性和溶出曲线、粒径等。当然我们也有不知道的数据,如参比制剂的粒径的数据,这些数据均有可能会影响药物在体内的吸收,我们决定采用z-factor模型用于代表不同晶型的体外溶出模型。
模型开发好之后,用软件中默认的餐后状态,进行不同晶型的模拟。该模型模拟结果为,食物能够促进药物吸收,即cmax和auc均有所提高,当然这也是bcsii类药物的共性,因为食物使胃肠道转运时间增加,增加了血流提及和药物的溶解度,更多的药物被溶解和吸收。
总结
上述研究中,建立pbpk模型预测仿制药和参比制剂食物的影响,并进行虚拟be,因不同晶型原料药性质不同,将不同原料药性质输入至模型中,如水溶性、粒径、体外溶出、fassif和fessif溶解性。这些参数也会影响餐后的模拟结果,我们必须要明智选择这些参数。
新领先研究模型建立
北京新领先医药科技有限公司拥有国内首个体内外桥接(ivivr)平台,以及文中提及的gastroplus软件,小编参考该文献数据,进行模拟,与文献对比值如下:
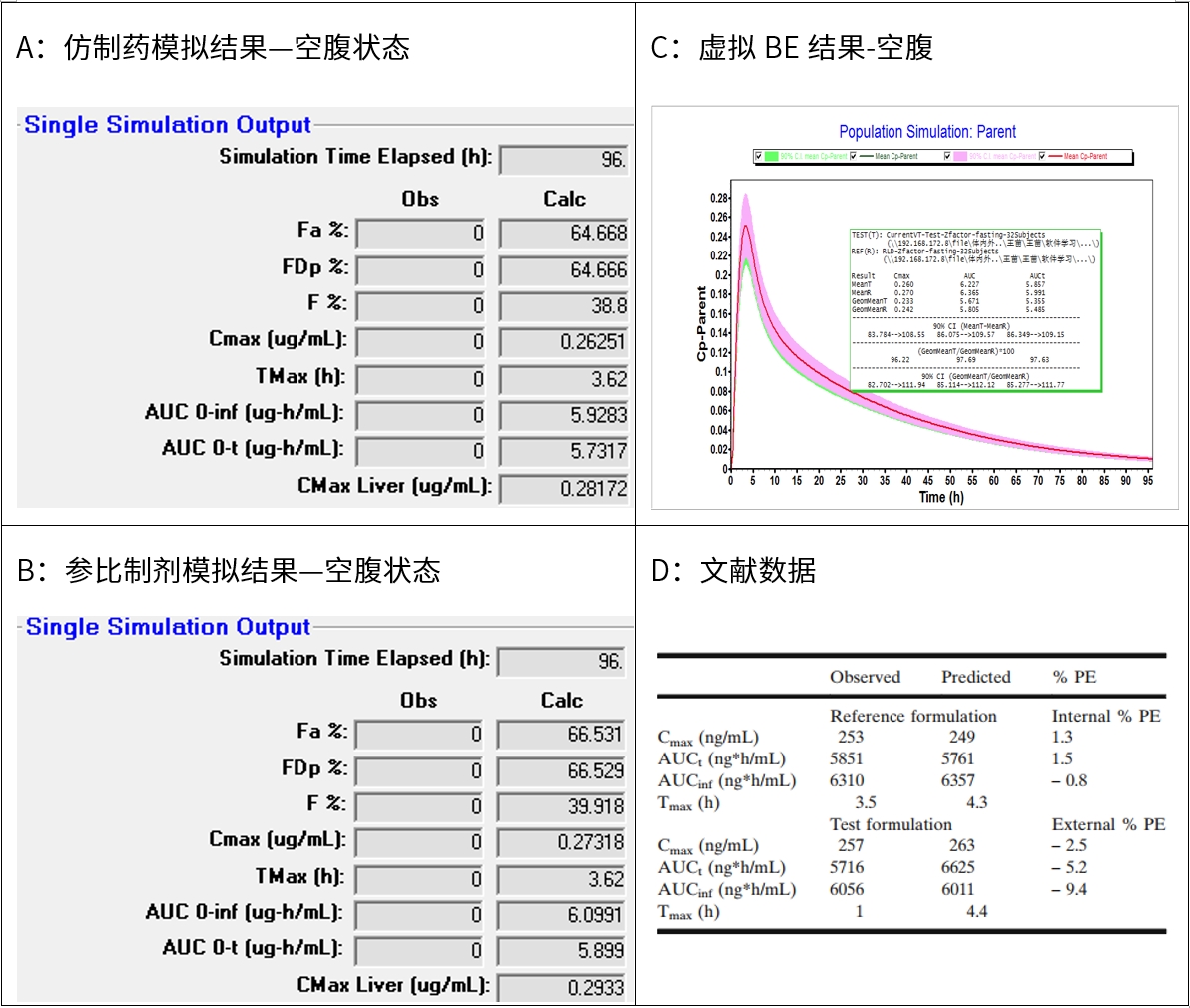
图5 新领先计算结果与文献结果对比-空腹状态
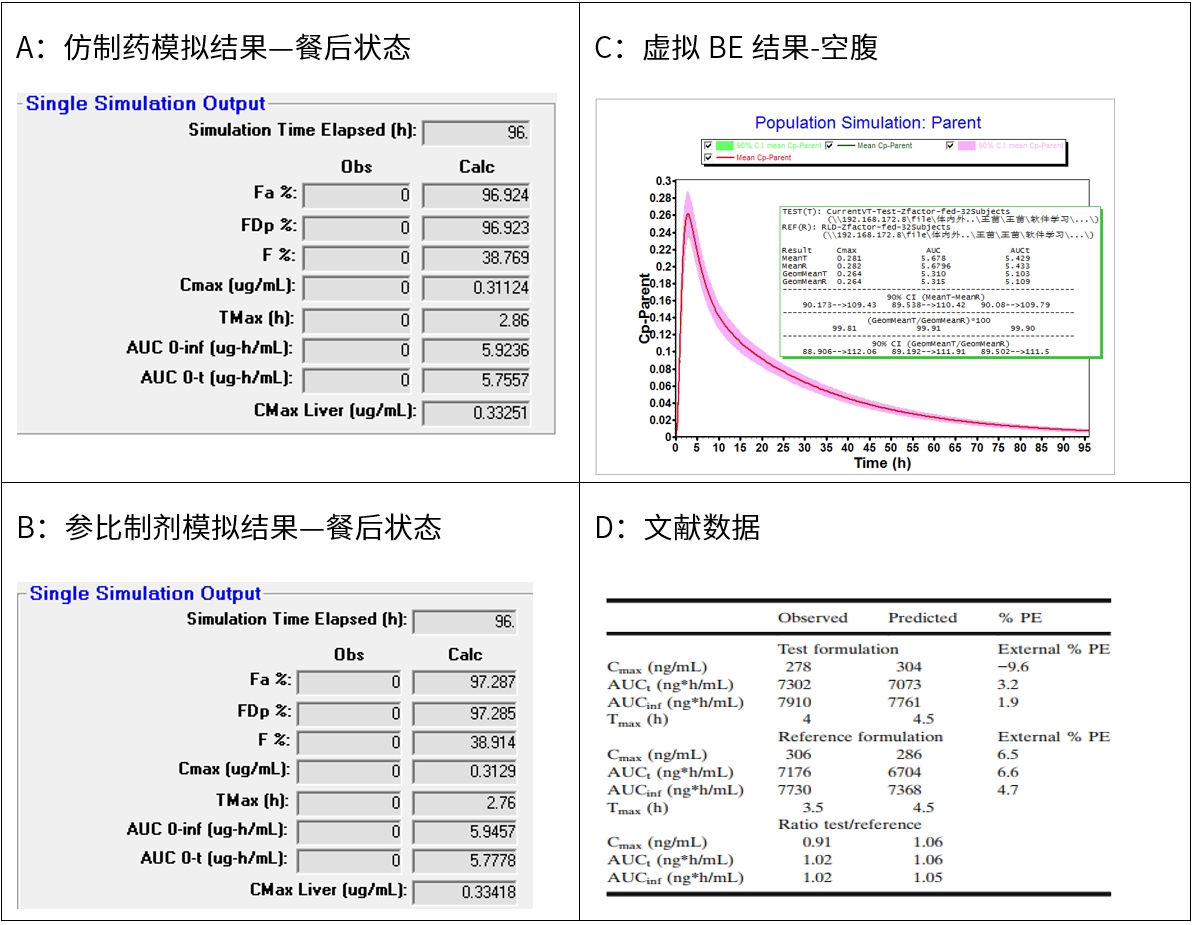
图6 新领先计算结果与文献结果对比-餐后状态
新领先参考文献值进行建模和模拟两个制剂的pk参数,cmax和auc采用单位分别为ug/ml和ug*h/ml,而文献中两个参数采用的单位分别为ng/ml和ng*h/ml,无论空腹和餐后,新领先预测cmax和auc与文献值报道无显著性差异。
因新领先无文献中提及的观测值,因此无法比较观测值和预测值的误差(pe)。文献中也提及预测值tmax较长,新领先模拟的结果也体现出tmax较长,该现象可能由于原料药溶解性较差,药物进入肠道中仍会有溶解和吸收。
参考文献数据,仿制药的z-factor数据为0.131,参比制剂z-factor值为0.205,模拟经验z-factor值越高,模拟结果越高,即参比吸收要比仿制药好,新领先模拟结果具有该趋势。可能由于模型中数据缺乏,我方模拟趋势与文献趋势相反。
无论空腹和餐后状态,仿制药和参比制剂虚拟be试验均生物等效,表明原料药晶型变更后,虽然原料药特性有所变化,但均能实现与仿制药生物等效。该模拟结果与文献报道一致。
-end-


转载声明:未经本网或本网权利人授权,不得转载、摘编或利用其他方式使用上述作品。已经本网或本网权利人授权使用作品的,应在授权范围内使用,并注明“来源:新领先医药科技”。

 010-61006450
010-61006450 联系地址:
联系地址: 技术市场部:
技术市场部: 北京新领先
北京新领先 新领先药讯
新领先药讯 010-61006450
010-61006450
